Los métodos de función de onda nacen con el desarrollo de la Química Cuántica, y están basados en la determinación de función de onda de un sistema desde primeros principios, ab initio. Una vez determinada la función de onda se pueden calcular diferentes propiedades del sistema en cuestión. Se podría decir tiene toda la información que necesitamos.
Los métodos empleados en química cuántica son muchos y variados, pero sin duda un paso fundamental fue el dado por Hartree en 1927 y posteriormente por Slater y Fock en los años 1929 y 1930, que finalmente dieron con la generalización del método de Hartree-Fock (HF). Desde entonces, y principalmente durante las décadas de los años 60 a 80, se han desarrollado diferentes procedimientos de cálculo y se han extendido estas aproximaciones a métodos que tienen en cuenta de mejor manera las contribuciones cuánticas a la correlación electrónica (los denominados métodos post- HF).
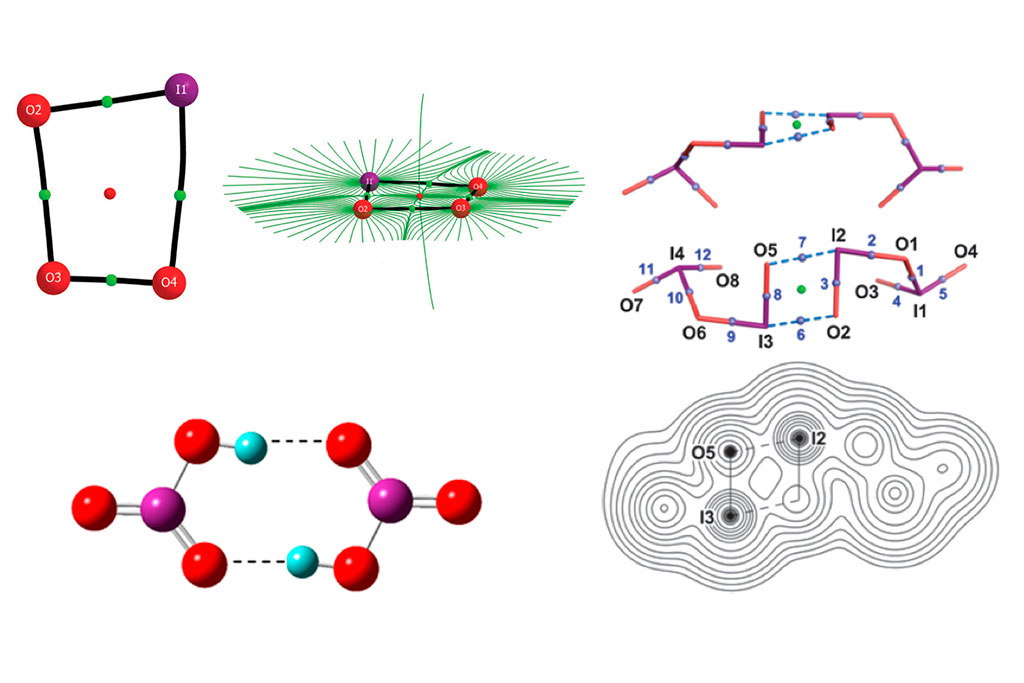
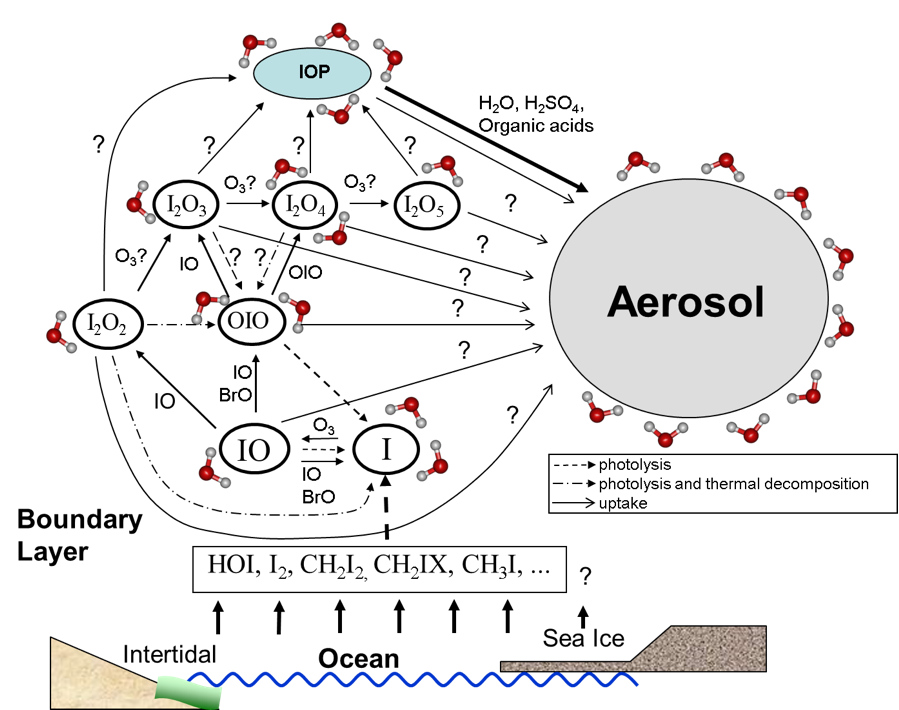
Desde el grupo de Modelling-UNED se emplean estos métodos para el cálculo de sistemas de interés atmosférico, a través de paquetes de cálculo ab initio como Gaussian o NWChem. A modo de ejemplo, estos métodos han sido empleados en anteriores ocasiones para la determinación de estructuras y caminos de reacción en los óxidos de yodo, responsables en mayor medida de los ciclos de eliminación de ozono en la troposfera.
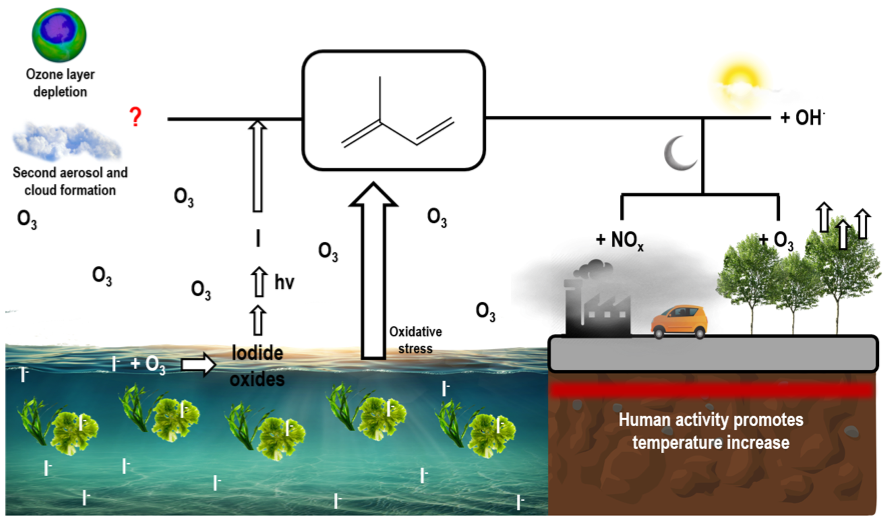
En la actualidad se realizan simulaciones encaminadas a conocer la estabilidad termodinámica así como la cinética de la molécula del isopreno y su interacción con átomos halógenos como el cloro, bromo y yodo.